

How is Retinitis Pigmentosa Inherited?
Inheritance
According to estimates, 100,000 Americans have RP, which is primarily brought on by changes (mutations) in a single gene that are inherited from either one or both parents. The mutant gene instructs photoreceptor cells to produce the erroneous protein, too little protein, or excessive protein. (To function effectively, cells require the right quantity of specific proteins.) Numerous gene mutations have been connected to RP.
One of three genetic transmission patterns—autosomal recessive, autosomal dominant, or X-linked—can transmit genetic changes from parent to child.
In autosomal recessive RP, neither parent exhibits any symptoms despite having one copy of the mutant gene and one normal copy in their bodies. They are referred to as unaffected carriers as a result. There is a 25% probability that each of their children will be impacted by receiving a mutant copy from each parent. The child will be an unaffected carrier if they only receive one copy of the mutation from one parent.
In autosomal dominant RP, often only one parent has a mutant copy of the gene and is the only parent who is affected. The inheritance of the modified copy has a 50% chance of having an impact on a kid. There is no evidence that the genetics of the unaffected parent contributes to the disease’s transmission.
The defective gene that causes X-linked RP is found on the X chromosome. Females have two X chromosomes, and one of them may carry the condition’s gene. Female carriers are less likely to develop X-linked disorders than non-carriers since they have a healthy copy of the gene on their other X chromosome. Males are genetically predisposed to X-linked disorders because they only have one X chromosome, which is paired with one Y chromosome. Because Y chromosomes are passed to sons by fathers who have X-linked disorders, their sons will never have an X-linked disease. A woman who has the X-linked disease gene has a 50% chance (or 1 in 2) of transferring the gene to her daughters, who subsequently become carriers, and a 50% chance of passing the gene to her sons, who subsequently develop the disease.
It is frequently suggested that if one family member has RP, the other family members should also undergo an eye check by a doctor who is highly qualified to check for retinal degenerative conditions. Discussing inheritability, family planning, genetic testing, and other relevant topics with a genetic counselor is a wise choice.
What is retinitis pigmentosa?
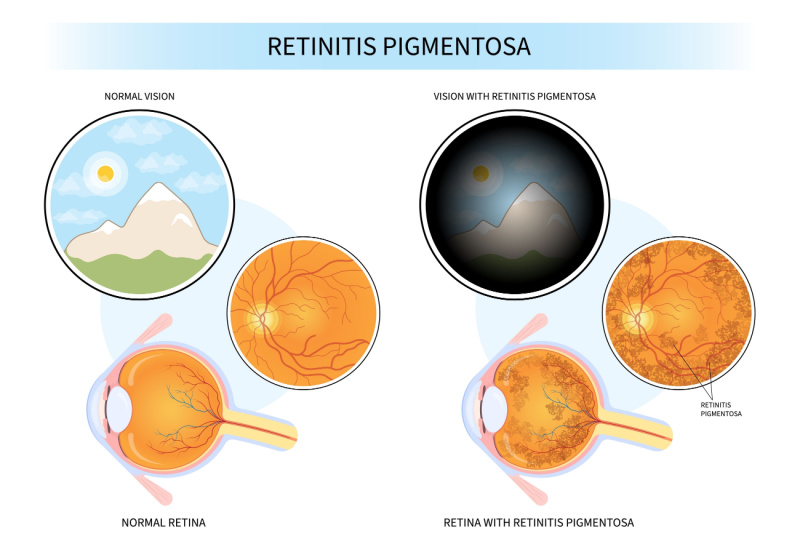
The term “retinitis pigmentosa” (RP) refers to a collection of inherited eye conditions that impair the retina, the portion of the eye that is sensitive to light. Photoreceptor cells, which are the retina’s light-detection cells, begin to deteriorate as a result of RP. We can see thanks to photoreceptor cells, which process and evaluate light. Patients gradually lose vision as these cells degenerate and die.
The gradual destruction of cones (retinal cells that perceive light and color) and rods (retinal cells that detect dim light) is the most typical aspect of all types of RP. Rod cells are first destroyed by the majority of RP types. These RP subtypes, also known as rod-cone dystrophy, typically start with night blindness. Night blindness resembles what normally sighted people go through when they enter a dark theater on a bright, sunny day. Patients with RP, however, struggle to adapt to surroundings that are gloomy and poorly lit.
The Two Types of Retinitis Pigmentosa
Depending on the relative involvement of rods and cones in the photoreceptors, there are two forms of retinitis pigmentosa. Early indications of Type 1 RP, which involves impaired night vision, include early and selective loss of rod sensitivity. Additionally, this illness has a slow rate of progression and results in localized visual loss.
Patients with Type 2 RP experience a progressive loss of both rod and cone sensitivity that starts in adulthood and is subsequently accompanied by deteriorating night vision.
Multiple Organ Involvement
When other organs are unaffected, retinitis pigmentosa might be nonsyndromic or “simple”; when other organs are damaged, it can be syndromic (example: Usher syndrome). X-linked inheritance, autosomal dominant inheritance, and autosomal recessive inheritance are all possible for nonsyndromic retinitis pigmentosa.
Digenic Inheritance
A rare digenic inheritance might also exist. People who have both ROM1 and RDS gene mutations exhibit digenic inheritance. Simplex instances are inherited via autosomal dominant, X-linked, or autosomal recessive mechanisms.
Retinitis pigmentosa genes and proteins
Proteins involved in the visual cycle, where the light reactive pigments are generated and recycled for functional vision, are encoded by genes linked to retinitis pigmentosa. Additionally, the transcription factors and proteins that make up the structure of photoreceptor cells are encoded by these genes.
Typical mutations
RHO gene mutations are a frequent occurrence. The rods are encoded by this gene, and mutations may cause RP, RP-associated congenital stationary night blindness, or, in rare cases, RP-associated autosomal recessive RP.
RDS mutations are also observed in a similar manner. Autosomal dominant RP, autosomal dominant macular degeneration, or digenic RP are caused by mutations in this gene, which typically codes for peripherin.
Here are a few prevalent genetic variants and the retinitis pigmentosa subtypes they are linked to:
Three gene mutations cause autosomal dominant retinitis pigmentosa, which is inherited as a dominant condition:
- RHO which is responsible for 25 to 30%
- RP1 – 5 to 10%
- RDS – 5 to 10%
There are more than 100 RHO mutations, and 10% of Americans with autosomal dominant retinitis pigmentosa frequently have P23H. Macular degeneration and complicated maculopathies may result from RDS mutations. The two most extensively researched RP1 mutations are Arg677stop and 2280del5.
These are more uncommon genetic mutations: autosomal recessive retinitis pigmentosa. Up to 5% of autosomal recessive instances may be attributable to mutations in USH2A, RPE65 (expressed in the RPE), PDE6A, and PDE6B.
X-Linked Retinitis Pigmentosa: In this kind, the RPGR (also known as RP3) and RP2 genes frequently exhibit alterations. They represent, respectively, 70–90% and 10–20% of all X-linked cases.
Mutations in the MT-TS2 are among the mitochondrial genes that cause non-syndromic retinitis pigmentosa.
Digenic retinitis pigmentosa is brought on by the presence of both an RDS gene mutation and a ROM1 gene mutation. Commonly, the RDS mutation is L185P.
Symptoms of Retinitis Pigmentosa
Patients experience tunnel vision loss as the condition worsens and more rod cells are lost. People with RP frequently have clear central vision but a ring of peripheral vision loss. Others say they have tunnel vision as if they are looking through a straw. Many retinitis pigmentosa individuals live their entire lives with some degree of central vision.
Other types of RP, also known as cone-rod dystrophy, initially impact central vision. Patients initially suffer from a loss of their center vision, which is unrecoverable with glasses or contacts. Changes in color perception accompany the loss of cone cells. Rod cells deteriorate as the condition worsens, resulting in night blindness and peripheral vision.
Children, teenagers, and young adults are the age groups where RP symptoms are most frequently noticed, and the condition continues to worsen throughout the patient’s lifetime. Variables include the pattern and severity of vision loss.
Retinitis Pigmentosa Treatment
Retinitis pigmentosa has no known treatment. The advancement of RP can be slowed down with a few treatment options, such as light avoidance and/or the use of low-vision devices. Some medical professionals think that vitamin A might be able to stop the advancement of RP. There is some evidence that taking high dosages of vitamin A (15,000 IU/day) may slightly halt development in some individuals. Vitamin A has relatively limited effects on disease and can be harmful if taken in excess. Before this type of therapy is widely recognized, more research needs to be done.
Additionally, research is being done on topics such as retinal prostheses, transplants, and gene therapy. Since RP typically results from a gene abnormality, gene therapy has drawn a lot of attention as a potential field of future study. Such studies would seek to understand how to introduce healthy genes into the retina. Experimental attempts to transplant healthy retinal cells into ill retinas are currently being made, but they have not yet been deemed clinically safe and effective. The function of a lost photoreceptor can be replaced with a prosthesis, a manufactured device meant to replace a broken body component, by electrically stimulating the retina’s remaining healthy cells. This makes retinal prosthesis a significant topic of research. The brain can receive a visual signal from the activated ganglion cells by electrical stimulation. A little decoder chip situated on the retinal surface receives the visual scene that a camera has recorded via electromagnetic radiation. The electrodes linked to the decoder are then provided with data and power. Individual electrodes provide electrical currents that excite retinal cells in the regions of the retina that correspond to the details in the visual picture.
Managing the Disease
People with RP and their families have access to a wide range of services, accommodating resources, and helpful tools. A low vision specialist can assist in making recommendations for the tools that are best for you.
Genetic Testing
For RP, genetic testing is available. It aids in determining the likelihood of a condition being passed from parent to child. It also aids in making a precise diagnosis. A patient is better able to decide which new therapy modalities and clinical trials are best for them when they have an accurate diagnosis.
Summary
A series of genetic eye conditions known as retinitis pigmentosa (RP) impair the retina, the area of the eye that receives light and transmits images to the brain. RP can be inherited in one of three ways: X-linked, autosomal dominant, or autosomal recessive.
A gene on one of the 22 pairs of autosomes that is responsible for autosomal dominant RP mutates. This indicates that the condition can be caused by a single copy of the defective gene. A 50% possibility exists that each kid of a person with autosomal dominant RP will inherit the faulty gene and experience the disorder.
Mutations in the same gene, which is present in two copies, one inherited from each parent, result in autosomal recessive RP. Although they do not themselves have the illness, the parents of a kid with autosomal recessive RP are carriers of the defective gene. There is a 25% probability that each child may inherit two copies of the defective gene and experience the condition if both parents are carriers.
Gene mutations on the X chromosome are the cause of X-linked RP. Males are more frequently affected by X-linked RP than females since males have one X and one Y chromosome whereas females have two X chromosomes. Males with X-linked RP receive the mutant gene from their mother and have a 50% chance of passing it on to their daughters, who will be carriers, and a 50% chance of passing it on to their sons, who will be affected by the condition.
To identify their risk of contracting the condition or passing it on to their offspring, family members of people with RP should think about genetic testing and counseling. Regular eye exams are essential for identifying any early indications of RP or other eye diseases. Family members should also be aware of the possible effects of RP on their everyday life and, as necessary, seek assistance from medical professionals and support organizations.
FAQ’s
Can retinitis pigmentosa skip a generation?
Retinitis Pigmentosa may skip generations even though it is inherited, making it challenging to trace. Any person who has a family member with a retinal condition ought to get checked out right away by an eye doctor.
Does retinitis pigmentosa affect women?
Female X-linked retinitis pigmentosa carriers can occasionally exhibit symptoms.
Are there any retinitis pigmentosa genes in the mother or father?
Most frequently, the ailment is caused by a recessive gene, which means that for the disease to manifest, the gene must be inherited from both parents. However, X chromosome genes and dominant genes have also been connected to retinitis pigmentosa. In some instances, the illness gene has only been transmitted from one parent.