Treatments for Retinitis Pigmentosa
A collection of connected eye conditions known as retinitis pigmentosa result in progressive vision loss. The retina, a layer of light-sensitive tissue in the back of the eye, is impacted by several diseases. Vision loss happens in retinitis pigmentosa patients when the retina’s light-sensing cells gradually degenerate.
The loss of night vision, which generally manifests in childhood, is the earliest indication of retinitis pigmentosa. It may be challenging to maneuver in low light if you have night vision issues. Later, the condition results in the development of blind spots in the peripheral (side) vision. These blind areas eventually converge to create tunnel vision. The condition worsens over years or decades, affecting central vision, which is essential for intricate tasks like reading, driving, and identifying people. Many adults with retinitis pigmentosa get legally blind at some point in their lives.
Most frequently, vision loss is the only retinitis pigmentosa symptom. Nonsyndromic refers to a condition that develops on its own. Nonsyndromic retinitis pigmentosa has been divided into three different subtypes, which are often defined by their mode of inheritance: autosomal dominant, autosomal recessive, or X-linked.
Retinitis pigmentosa develops less often as a result of disorders that affect other body organs and tissues. These illness manifestations are referred to as syndromic. Usher syndrome, the most prevalent kind of syndromic retinitis pigmentosa, is characterized by the onset of visual and hearing loss simultaneously at a young age. Other hereditary syndromes with retinitis pigmentosa as a characteristic include Bardet-Biedl syndrome, Refsum disease, neuropathy, ataxia, and retinitis pigmentosa (NARP).
What do we know about retinitis pigmentosa and heredity?
Since RP is a hereditary condition, it could also impact a different family member. Retinal cells depend on a variety of particular genes to produce vision since they are among the most specialized cells in the human body. Loss of eyesight may result from a disease-causing mutation in any one of these genes. Over 100 genes have been identified by researchers as having potential mutations that cause retinitis pigmentosa. Most RP cases—about 50%—are solitary and have no prior family history. There is no known reason for these cases. Other RP instances with known family histories can be classified as autosomal recessive, autosomal dominant, or X-linked recessive.
When both parents are asymptomatic carriers of the same faulty gene, autosomal recessive RP develops. One in four children will likely be impacted. This implies that both parents must pass on the faulty gene to the afflicted kid. One in two parents had unaffected children who would be carriers of the problematic gene. One in four parents will have a child who is fully devoid of the RP gene.
Only when one copy of the gene is faulty in autosomal dominant RP does the condition affect either males or females. Usually, one of the parents will have the illness. If the sick parent carries both a normal and a faulty gene, there is a one in two probability that any given offspring will get the condition.
Offspring may get X-linked recessive RP in one of two ways. Mothers may have faulty genes or the men may be afflicted. All sons will be unaffected and all girls will be carriers if the father has the condition. One in two sons will be afflicted and one in two girls will be a carrier if the mother is the carrier. Only males in families with the X-linked type have severe vision loss; females possess the genetic characteristic but do not.
What causes Retinitis Pigmentosa?
Nonsyndromic retinitis pigmentosa is thought to be caused by mutations in more than 60 genes. The autosomal dominant type of illness is linked to more than 20 of these genes. Twenty to thirty percent of all instances of autosomal dominant retinitis pigmentosa are caused by mutations in the RHO gene. The autosomal recessive version of the condition has at least 35 genes linked to it. The most prevalent of them is USH2A; 10–15% of all instances of autosomal recessive retinitis pigmentosa are caused by mutations in this gene. The X-linked type of illness is considered to be brought on by changes in at least six genes. The majority of instances of X-linked retinitis pigmentosa are caused by mutations in the RPGR and RP2 genes together.
The shape and operation of the retina’s specialized light receptor cells, or “photoreceptors,” are significantly influenced by the genes linked to retinitis pigmentosa. Rods and cones, two different kinds of photoreceptors, are found in the retina. While cones are in charge of vision in bright light, including color vision, rods are in charge of seeing in low light.
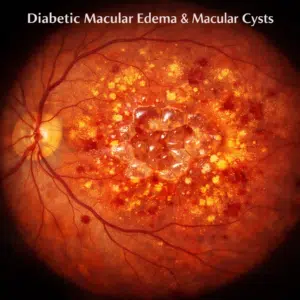
The rods and cones in the retina gradually disappear as a result of mutations in any of the retinitis pigmentosa-causing genes. The distinctive pattern of vision loss seen in those with retinitis pigmentosa is brought about by the gradual degradation of these cells. Because rods degrade before cones do, night vision impairment is frequently the initial symptom of the condition. Later, when both rods and cones are eliminated, daytime vision becomes impaired.
Some of the genes linked to retinitis pigmentosa are also linked to other conditions affecting the eyes, such as cone-rod dystrophy. The signs and symptoms of cone-rod dystrophy are comparable to those of retinitis pigmentosa. However, because the cones in cone-rod dystrophy deteriorate before the rods, daytime and color vision are impacted before night vision.
What is the frequency of retinitis pigmentosa?
Retinitis pigmentosa is thought to affect 1 in 3,500 to 1 in 4,000 persons in Europe and the US. Around two million people worldwide, or 1 in 3,000 to 1 in 4,000 individuals, are affected by RP. This number is thought to be around 100,000 persons in the United States.
Retinitis pigmentosa signs and symptoms
Retinitis pigmentosa symptoms often appear in infancy or adolescence. However, every individual may experience symptoms in a unique way. Some affected individuals have a gradual, rapid loss of vision. Some people experience significantly more severe and rapid vision loss. Typical signs could include:
- Having trouble seeing in low light or the dark
- A diminished capacity for either central, side, or peripheral vision
- Difficulty reading print
- Los of detailed vision
- Having trouble stumbling or falling over items that are not visible
- Glare
The condition’s symptoms might resemble those of other eye conditions. Consult your eye care professional for a diagnosis.
Retinitis pigmentosa therapies currently available
Retinitis pigmentosa patients can presently choose between two therapies.
Correcting RPE65 gene mutations
Only individuals with mutations in both copies of the RPE65 gene are eligible for the gene therapy Luxturna. This mutation impairs the retina’s ability to react to light. A normal copy of the RPE65 gene is directly delivered to the retina by a single injection of Luxturna. The retina’s capacity to react to light is restored as a result.
Luxturna can enhance night vision and aid patients in better navigating in low light if they take the medication soon enough after diagnosis.
Spark Therapeutics created Luxturna, which the US Food and Drug Administration authorized in 2017. Both eyes cost $850,000, which your insurance company could cover.
Advanced Treatments for Retinitis Pigmentosa
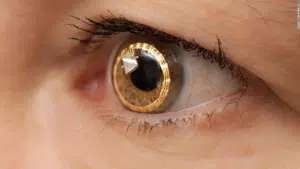
Using the Argus II bionic eye, patients with advanced retinitis pigmentosa could see a little improvement in their vision. Patients that respond well to this therapy frequently have very poor vision and may only be able to detect light. A tiny electronic gadget has to be surgically implanted on the retina. Additionally, patients are required to wear a certain set of glasses equipped with video processing equipment. Pictures from the glasses are sent to the apparatus, which in turn activates the retina’s light-sensitive cells and sends the images to the brain.
The updated spectacles for compatibility with previously implanted Argus II devices were introduced by Second Sight Medical Products earlier in 2021, but they have not yet reached the market. An alternative implant is being developed in Australia even though additional Argus II implants are no longer sold there.
Future treatments for retinitis pigmentosa are being developed.
People with RP are expected to benefit from a number of novel therapies. Many of these treatments are still undergoing testing, so it will probably be years before patients can access them. However, the early results are encouraging. If you think you could be eligible for a clinical trial, ask your ophthalmologist.
A potential treatment for all forms of retinitis pigmentosa
No matter what their genetic mutation is, persons with retinitis pigmentosa may benefit from the therapy GenSight Therapeutics is studying. Optogenetic therapies are treatments that employ light to manipulate cells.
An eye injection and the usage of cutting-edge goggles are combined in the optogenetic treatment offered by GenSight. A gene that helps retinal cells react to light is delivered through injection. These cells can communicate with the brain electrically thanks to the goggles. The injection and the goggles try to mimic the function of photoreceptors, which are light-sensing cells that don’t function properly in persons with retinitis pigmentosa.
To master the usage of eyewear, patients must train for several months. Five people have received the therapy thus far. Some people have improved their capacity to recognize crosswalk lines on the ground and discern high-contrast things on a table. They are still unable to read, identify faces, or operate a vehicle. Retrosense Therapeutics (Allergan), Nanoscope Therapeutics, and Vedere Bio, Inc. are further organizations working on optogenetic treatments (Novartis).
X-linked retinitis pigmentosa gene therapy
Three firms, Meira GTX, Applied Genetic Technologies, and BioGen, are working on experimental gene treatments that may help people with the aggressive type of X-linked retinitis pigmentosa. Men are more frequently affected by this illness, which is brought on by an RPGR gene mutation.
A surgery called a vitrectomy and an injection into the eye that distributes healthy copies of the RPGR gene to the macula, a region of the retina, are used in all three therapies. Clinical study participants who had treatment in one eye reported improvements in their visual field, light sensitivity, and capacity for navigating in low light. Another kind of gene therapy without a vitrectomy is being tested by a business by the name of 4D Molecular Therapeutics.
Repairing the USH2A gene’s defects
A gene treatment being developed by ProQR Therapeutics may prevent vision loss in persons with Usher syndrome and retinitis pigmentosa caused by a mutation in the USH2A gene. Patients who have this mutation are unable to produce the USH2A protein, which is necessary for eyesight. The treatment, known as QR-421a, is injected into the eye and enables cells to make a more beneficial USH2A protein. So far, both visual acuity and visual field have improved in individuals with advanced and early-moderate illnesses. In the fall of 2021, ProQR Therapeutics plans to test the treatment in phase 2-3 clinical study.
Repairing the RHO gene’s defects
Therapy for patients with retinitis pigmentosa caused by a mutation in the RHO gene is being tested in different research by ProQR Therapeutics. This also goes by the name RP4. A defective form of the rhodopsin protein, which typically transforms light into an electrical signal, is produced by people who have this mutation. Over time, the retina is poisoned by the defective protein. The brand-new medication, QR-1123, works by stopping the production of the defective protein and is administered through ocular injection. This enables the protein’s regular form to once more control the retina. A phase 1-2 clinical trial is now investigating this treatment.
Treating type 10 Leber congenital amaurosis
Infants who have Leber congenital amaurosis suffer from retinitis pigmentosa, a kind of disease. The retina’s light-sensing cells are destroyed by this illness. A CEP290 gene deficiency that results in type 10 disease and, frequently, legal blindness causes gradual vision loss.
For this therapy, there are two potential treatments under research.
CRISPR is a gene-editing technique that scientists have created to attempt to fix genetic mutation. During an eye injection, the retina receives the therapy. The tool is intended to assist the retina in producing a protein that both delays cell death and revives certain already deceased cells. This could aid in patients’ visual recovery. The phase 1-2 BRILLIANCE clinical trial, including 18 participants, is being run by Allergan and Editas Medicine.
RNA antisense oligonucleotide treatment is a sort of gene therapy that ProQR Therapeutics is exploring. This therapy allows for the creation of a protein required for eyesight and is administered via ocular injection. Several months after injection, patients in phase 2-3 clinical trials report remarkable advancements in their eyesight and retinal structure.
These therapies may be able to prevent people from suffering from retinitis pigmentosa’s crippling vision loss.
For some types of retinitis pigmentosa, gene therapy offers the chance to stop the otherwise unrelenting loss of eyesight and visual field. It may even help some individuals see better, be more sensitive to light, and be able to see and navigate in the dark.
FAQ’s
Do people with retinitis pigmentosa entirely go blind?
It should be remembered that RP is a condition that develops slowly over many years, and the majority of sufferers never entirely lose their vision. Even while many persons with RP are regarded as “legally blind,” this is only true because their fields of vision are extremely limited (poor peripheral vision).
What vitamin aids in the treatment of retinitis pigmentosa?
Vitamin A and fish oils have been shown to be effective and safe in slowing the course of vision loss in persons with retinitis pigmentosa (RP).
Can retinitis pigmentosa be stopped?
There is currently no recognized treatment for retinitis pigmentosa, nor can the damage caused by this disorder be undone. The advancement of retinitis pigmentosa, however, may be slowed down, allowing patients to keep their eyesight for a longer period of time, according to some of the most recent studies.
What age does retinitis pigmentosa cause blindness?
It is well known that some people with retinitis pigmentosa are nearly blind by the age of 30, while others are able to see well into their 80s or beyond. Even among individuals in the same family, there can be a significant variation in the severity of the disease at a particular age.